
FastQC A Quality Control tool for NGS. About | People | Services | Projects | Training | Publications FastQC View our tutorial video FastQC aims to provide a simple way to do some quality control checks on raw sequence data coming from high throughput sequencing pipelines.
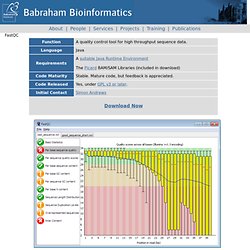
It provides a modular set of analyses which you can use to give a quick impression of whether your data has any problems of which you should be aware before doing any further analysis. The main functions of FastQC are Import of data from BAM, SAM or FastQ files (any variant) Providing a quick overview to tell you in which areas there may be problems Summary graphs and tables to quickly assess your data Export of results to an HTML based permanent report Offline operation to allow automated generation of reports without running the interactive application Documentation A copy of the FastQC documentation is available for you to try before you buy (well download..).
Example Reports Changelog. Illumina Quality Scores for NGS. Phred quality score - What is it ? Phred quality scores shown on a DNA sequence trace Phred quality scores were originally developed by the program Phred to help in the automation of DNA sequencing in the Human Genome Project.
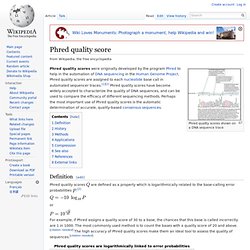
Phred quality scores are assigned to each nucleotide base call in automated sequencer traces.[1][2] Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods. Perhaps the most important use of Phred quality scores is the automatic determination of accurate, quality-based consensus sequences.
Definition[edit] Phred quality scores are defined as a property which is logarithmically related to the base-calling error probabilities or For example, if Phred assigns a quality score of 30 to a base, the chances that this base is called incorrectly are 1 in 1000. The phred quality score is the negative ratio of the error probability to the reference level of expressed in Decibel (dB). History[edit] Bowtie: An ultrafast, memory-efficient short read aligner.
TopHat - fast splice junction mapper for RNA-Seq reads. TopHat 2.1.1 release 2/23/2016 Please note that TopHat has entered a low maintenance, low support stage as it is now largely superseded by HISAT2 which provides the same core functionality (i.e. spliced alignment of RNA-Seq reads), in a more accurate and much more efficient way.

Version 2.1.1 is a maintenance release which includes the following changes, some of them thanks to GitHub contributors: TopHat can be now built on more recent Linux distributions with newer GNU C++ (5.x), as the included SeqAn library was finally upgraded to a newer version. Improved the detection of linker options for the Boost::Thread library which prevented the TopHat build from source on some systems. Incorporated Luca Venturini's code to support large bowtie2 indexes (.bt2l). TopHat 2.1.0 release 6/29/2015 TopHat-Fusion algorithm improvements for more sensitive and accurate discovery of fusions, thanks to contributions from Gordon Bean and Ryan Kelley at Illumina. TopHat 2.0.14 release 3/24/2015. Cufflinks - Transcript assembly, differential expression, and differential regulation for RNA-Seq.
SeqMonk Mapped Sequence Analysis Tool (viewer) Integrative Genomics Viewer. Strand-ngs. Using R for Sequence Analysis. Bioconductor can import diverse sequence-related file types, including fasta, fastq, BAM, gff, bed, and wig files, among others.
Packages support common and advanced sequence manipulation operations such as trimming, transformation, and alignment. Domain-specific analyses include quality assessment, ChIP-seq, differential expression, RNA-seq, and other approaches. Bioconductor includes an interface to the Sequence Read Archive (via the SRAdb package). Sample Workflow. Chipster. Genomatix Genome Analyzer - Genomatix. The Genomatix Genome Analyzer (GGA) is our integrated solution for comprehensive visualization and interpretation of Next Generation Sequencing (NGS) data from ChIP, RNA, DNA, methylation or small RNA sequencing.

Each analyzer is brimming with state-of-the-art technology that sheds light on biological context – essential to help you understand the big picture. The GGA produces results of higher relevance, answering your scientific questions with greater precision than ever before. The biological background data consisting of annotation and gene network data provided by ElDorado plus the transcription factor knowledge contained in MatBase lets researchers analyze and interpret their experimental results in a unique biological context on every GGA for more than 30 different species.
Combined with the Genomatix Mining Station the Genomatix Genome Analyzer provides a complete analysis solution from raw sequencing tags to the biology behind your data. CLC Genomics Workbench - CLC bio. Dominating the high-throughput sequencing dataanalysis challenge We have overcome the challenge to analyze high-throughput sequencing data faster than it is produced by implementing a SIMD-accelerated assembly algorithm in our next generation sequencing solution, CLC Genomics Workbench – a cross-platform desktop application with a graphical user-interface.
CLC Genomics Workbench, for analyzing and visualizing next generation sequencing data, incorporates cutting-edge technology and algorithms, while also supporting and integrating with the rest of your typical NGS workflow. CLC Genomics Workbench is available for Windows, Mac OS X, and Linux platforms. It includes a number of features within the fields of genomics, transcriptomics and epigenomics, and additionally it includes all the tools of CLC Main Workbench.