
PII: S0065-3233(01)55002-0 - 10_AdvProtChem.pdf. 09Matthews_Itzhaki.pdf. dConsensus: a tool for displaying domain ... [BMC Bioinformatics. 2010. Prototypes of elementary functional loops unravel evolutionary connections between protein functions. + Author Affiliations * To whom correspondence should be addressed.

Abstract Motivation: Earlier studies of protein structure revealed closed loops with a characteristic size 25–30 residues and ring-like shape as a basic universal structural element of globular proteins. Elementary functional loops (EFLs) have specific signatures and provide functional residues important for binding/activation and principal chemical transformation steps of the enzymatic reaction. The goal of this work is to show how these functional loops evolved from pre-domain peptides and to find a set of prototypes from which the EFLs of contemporary proteins originated. Results: This article describes a computational method for deriving prototypes of EFLs based on the sequences of complete genomes.
Contact: Igor.Berezovsky@uni.no Enzymes are involved in all processes in living organisms. The first question that arises in this context is what elements of protein folds serve as elementary units of function. DOMIRE: a web server for identifying structural domains and their neighbors in proteins. + Author Affiliations ↵* To whom correspondence should be addressed.

Received June 16, 2011. Revision received January 19, 2012. Accepted February 8, 2012. Abstract. A thermodynamic definition of protein domains. Author Affiliations Edited by* S.

Walter Englander, University of Pennsylvania, Philadelphia, PA, and approved April 10, 2012 (received for review February 13, 2012) Abstract Protein domains are conspicuous structural units in globular proteins, and their identification has been a topic of intense biochemical interest dating back to the earliest crystal structures. Numerous disparate domain identification algorithms have been proposed, all involving some combination of visual intuition and/or structure-based decomposition. Domains are visually arresting protein substructures with an influential history in protein biochemistry (1). Dissecting proteins into their constituent domains provides a simple, intuitive approach to classifying protein structure, a molecular application of the time-honored principle of “carving nature at its joints” (5).
However, seeing can be deceiving. Cited In for PubMed (Select 9521098) MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Abstract Motivation: Molecular recognition features (MoRFs) are short binding regions located within longer intrinsically disordered regions that bind to protein partners via disorder-to-order transitions.

MoRFs are implicated in important processes including signaling and regulation. However, only a limited number of experimentally validated MoRFs is known, which motivates development of computational methods that predict MoRFs from protein chains. Results: We introduce a new MoRF predictor, MoRFpred, which identifies all MoRF types (α, β, coil and complex). We develop a comprehensive dataset of annotated MoRFs to build and empirically compare our method. Availability: Contact: lkurgan@ece.ualberta.ca Supplementary information: Supplementary data are available at Bioinformatics online. An analysis of protein domain linkers: their classification and role in protein folding. Introduction Many cellular processes involve proteins with multiple domains.

The modular nature of proteins has many advantages, providing increased stability and new cooperative functions. Other advantages include the protection of intermediates within inter-domain clefts that may otherwise be unstable in aqueous environments and the fixed stoichiometric ratio of enzymatic activity necessary for a sequential set of reactions (Ostermeier and Benkovic, 2000).
It is not surprising then that advances in protein engineering have come from creating multi-functional chimeric proteins containing modules from various proteins (e.g. Nixon et al., 1997). Recent studies have shown that domain linkers can play an essential role in maintaining cooperative inter-domain interactions (for a review see Gokhale and Khosla, 2000). Some previous studies have identified particular types of linker. Methods. Online Analysis Tools - Motifs. Motif Scan – (MyHits, SIB, Switzerland) includes Prosite, Pfam and HAMAP profiles.

InterPro 5 - includes PROSITE, HAMAP (High-quality Automated and Manual Annotation of Proteins), Pfam (protein Families), PRINTS, ProDom, SMART (a Simple Modular Architecture Research Tool), TIGRFAMs, PIRSF (Protein Information Resource), SUPERFAMILY, CATH-Gene3D (Class, Architecture, Topology, Homologous superfamily), and PANTHER (Protein ANalysis THrough Evolutionary Relationships) classification systems. (Reference: Jones, P. et al. 2014, Bioinformatics 10: 1093) . This service is also available here. MOTIF (GenomeNet, Japan) - I recommend this for the protein analysis, I have tried phage genomes against the DNA motif database without success. Protein domain. Pyruvate kinase, a protein with three domains.
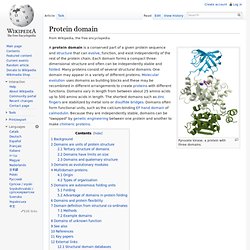
Background[edit] The concept of the domain was first proposed in 1973 by Wetlaufer after X-ray crystallographic studies of hen lysozyme [1] and papain [2] and by limited proteolysis studies of immunoglobulins.[3][4] Wetlaufer defined domains as stable units of protein structure that could fold autonomously. In the past domains have been described as units of: compact structure[5]function and evolution[6]folding.[7] Each definition is valid and will often overlap, i.e. a compact structural domain that is found amongst diverse proteins is likely to fold independently within its structural environment.
An appropriate example is pyruvate kinase, a glycolytic enzyme that plays an important role in regulating the flux from fructose-1,6-biphosphate to pyruvate. The central α/β-barrel substrate binding domain is one of the most common enzyme folds.