
Sebastian Kurscheid: RNA-S... Seq Tutorial from the iPlant Collaborative. Sebastian Kurscheid: Tutor... Tutorial on RNA-seq mapping and data analysis. Sebastian Kurscheid: RNA-S... Sebastian Kurscheid: RNA-S... Seq Data Analysis – Poll Results. Sebastian Kurscheid: RNA A... RNA Advances Reshape Prevailing Wisdom. From Genetic Engineering News Expanding on a concept originally coined by Walter Gilbert in 1986, Thomas Cech recently described two RNA worlds—a hypothetical, primordial world in which the same molecule combined informational and catalytic properties, and a contemporary world, forged by a spectrum of RNA-centered activities.
While during the early days, following the discovery of DNA, RNA was thought to be the less interesting molecule, this view has dramatically changed in recent years. Advances in RNA biology are reshaping concepts that have been prevailing for a long time. For example, in the field of developmental biology, decisions were traditionally envisioned mostly in terms of the progressive expression of different combinations of transcription factors that are induced by specific combinations and concentrations of growth differentiation factors.
(read more…) Stein, RA. (2012) RNA Advances Reshape Prevailing Wisdom. Sebastian Kurscheid: RNA-s... Seq Workshop. Sebastian Kurscheid: Oases... Sebastian Kurscheid: RNA-S... RNA-Seq Data Analysis – Where To Start? In our last reader poll, we asked: Do we yet have a firm handle on the most appropriate/accurate pipeline for analysis of RNA-Seq datasets?
The overwhelming result was: NO. N=72 We’re hearing feedback now from scientists developing data analysis methods for RNA-Seq. Would very much like to hear from more of you. Please send your comments to contribute@rna-seqblog.com. by Dr. Concerning the poll results I perfectly understand the frustration of the researchers. Although RNA-seq seems to be an extremely powerful technique, only now we are getting information on bias and criticality. In my opinion a keyword for pipeline optimization is “benchmark dataset”. On the basis of previous works, it is evident that we desperately need experimental benchmark datasets to optimize RNA-seq pipelines. Sebastian Kurscheid: RNA-S... RNA-Seq Data Analysis Workshop. Sebastian Kurscheid: RNA-S... RNA-Seq Workshops.
Sebastian Kurscheid: Model... Modeling RNA degradation for RNA-Seq. Methods for the estimation of the transcript’s abundance using RNA-Seq data have been intensively studied, many of which are based on the assumption that the short-reads of RNA-Seq are uniformly distributed along the transcripts.
However, the short-reads are found to be nonuniformly distributed along the transcripts, which can greatly reduce the accuracies of these methods based on the uniform assumption. Several methods are developed to adjust the biases induced by this nonuniformity, utilizing the short-read’s empirical distribution in transcript. Researchers at USC and the Chinese Academy of Sciences have found that RNA degradation plays a major role in the formation of the short-read’s nonuniform distribution and thus developed a new approach that quantifies the short-read’s nonuniform distribution by precisely modeling RNA degradation. They showed that their method can improve the accuracy of transcript isoform expression estimation. Sebastian Kurscheid: RNA-S... RNA-Seq to Measure Protein Abundance. Within the proteomics community there is a substantial interest in development of novel label-free quantitative proteomic strategies.
One strategy is to take advantage of an increasing number of studies involving integrative analysis of gene and protein expression data that are based on new technologies such as next-generation transcriptome sequencing (RNA-Seq) and highly sensitive mass spectrometry (MS) instrumentation. Thus, it becomes interesting to revisit the correlative analysis of gene and protein expression data using more recently generated datasets to determine if gene expression data can be used as an indirect benchmark for such protein-level comparisons. A team of researchers from the University of Michigan and the Chinese Academy of Sciences used publicly available mouse data to perform a joint analysis of genomic and proteomic data obtained on the same organism.
Sebastian Kurscheid: Lever... Leveraging in Statistical Analysis of RNA-Seq. SPEAKER: Professor Ping Ma Department of Statistics Univ. of Illinois at Urbana-Champaign TIME & PLACE: Monday, February 20, 2012 12:00-1:00p The Forum, WID/MIR Building Dept of Statistics Univ of Wisconsin, Madison ABSTRACT: With the rapid development of second-generation sequencing technologies, RNA-Seq has become a popular tool for transcriptome analysis.
It offers the chance to detect novel transcripts by obtaining tens of millions of short reads. After mapped to the genome and/or to the reference transcripts, RNA-Seq data can be summarized by a tremendous number of short-read counts. Sebastian Kurscheid: Compr... Comprehensive profile of the RNA editome. A Team of researchers from Taiwan & Denmark have performed a comprehensive profiling of the RNA editome of a male Han Chinese individual based on analysis of ~767 million sequencing reads from poly(A)+, poly(A)− and small RNA samples.
Developed a computational pipeline that carefully controls for false positives while calling RNA editing events from genome and whole-transcriptome data of the same individual.Identified 22,688 RNA editing events in noncoding genes and introns, untranslated regions and coding sequences of protein-coding genes. Most changes (~93%) converted A to I(G), consistent with known editing mechanisms based on adenosine deaminase acting on RNA (ADAR).Found evidence of other types of nucleotide changes; however, these were validated at lower rates.Found 44 editing sites in microRNAs (miRNAs), suggesting a potential link between RNA editing and miRNA-mediated regulation. Sebastian Kurscheid: A Sim... Sebastian Kurscheid: RNA-S... Seq Poll Results. Sebastian Kurscheid: Last... Last Month’s RNA-Seq Industry Press. Sebastian Kurscheid: RNA-S... Sebastian Kurscheid: A new...
Sebastian Kurscheid: RNA-S... Seq of Cows Milk. Cow milk is a complex bioactive fluid consumed by humans beyond infancy.
Even though the chemical and physical properties of cow milk are well characterized, very limited research has been done on characterizing the milk transcriptome. This study performs a comprehensive expression profiling of genes expressed in milk somatic cells of transition (day 15), peak (day 90) and late (day 250) lactation Holstein cows by RNA sequencing.
Milk samples were collected from Holstein cows at 15, 90 and 250 days of lactation, and RNA was extracted from the pelleted milk cells. Gene expression analysis was conducted by Illumina RNA sequencing. Sequence reads were assembled and analyzed in CLC Genomics Workbench. The results revealed that 69% of NCBI Btau 4.0 annotated genes are expressed in bovine milk somatic cells. Wickramasinghe S, Rincon G, Islas-Trejo A, Medrano JF. (2012) Transcriptional profiling of bovine milk using RNA sequencing. Sebastian Kurscheid: FX: a... Sebastian Kurscheid: RNA S... Sebastian Kurscheid: Revie... Review of RNA-Seq Data Analysis Software Tools. This review summarizes a number of frequently-used applications of transcriptome sequencing and their related analyzing strategies, including short read mapping, exon-exon splice junction detection, gene or isoform expression quantification, differential expression analysis and transcriptome reconstruction.
Table 1 Tools for short read mappingTable 2 A list of software for splice junction detectionTable 3 Software for gene or isoform expression quantificationTable 4 Available tools for differential expression analysisTable 5 Transcriptome reconstruction tools. Sebastian Kurscheid: Truly... Truly Digital RNA-Seq. Researchers at Harvard University have developed a method of truly digital RNA-Seq that does not rely on the counting of individual reads.
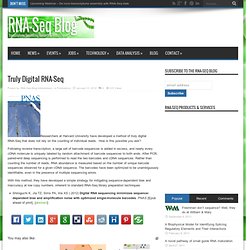
How is this possible you ask? Following reverse transcription, a large set of barcode sequences is added in excess, and nearly every cDNA molecule is uniquely labeled by random attachment of barcode sequences to both ends. After PCR, paired-end deep sequencing is performed to read the two barcodes and cDNA sequences. Rather than counting the number of reads, RNA abundance is measured based on the number of unique barcode sequences observed for a given cDNA sequence. The barcodes have been optimized to be unambiguously identifiable, even in the presence of multiple sequencing errors. With this method, they have developed a simple strategy for mitigating sequence-dependent bias and inaccuracy at low copy numbers, inherent to standard RNA-Seq library preparation techniques.
Sebastian Kurscheid: The C... The Challenge of RNA-Seq. The authors of a recent review1, make some key points about some of the challenges that are general to all RNA-seq experiments: RNA-Seq rules!
– “By comparison to these [other next-gen sequencing] applications, RNA-sequencing (RNA-seq) may be leading the pack in popularity because of its ability to characterize transcriptomes, to assess differential gene expression and to essentially challenge the continued use of microarray technology for studying transcription.” There is no magic to RNA-Seq. – “We are concerned that next-generation technologies will, most likely, remain expensive for a while, and that there may be an inclination to revert to single sample science that is void of any ability to estimate biological and/or technical variation, or to test scientific hypotheses.” Sebastian Kurscheid: GC-Co... Sebastian Kurscheid: Diffe...